Materials and Methods
⁎ Materials
All chemicals were used without further purification. Lipase from C. antarctica B was provided by Novozymes. Graphite oxide was supplied by Graphene Company (Turkey). Ferrous chloride tetrahydrate (FeCl2. 4H2O, 99%), ferric chloride hexahydrate (FeCl3. 6H2O, 97%), glutaraldehyde (GA, %50 wt. in H2O), epichlorohydrin (EPH), and Triton X-100 were obtained from Sigma-Aldrich. Calcium chloride (CaCl2), sodium hydroxide (NaOH), toluene, hexane, 1-phenyl ethanol, 2-propanol, vinyl acetate, hydrochloric acid (HCl), tween 80, disodium hydrogen phosphate dihydrate (Na2HPO4.2H2O), and monopotassium phosphate (KH2PO4) were purchased from Merck.
⁎ Synthesis of Nanostructures
⭒ Synthesis of Fe3O4 Nanoparticles
Fe3O4 nanoparticles were prepared by coprecipitation method in alkaline medium reported method in the literature [39]. The iron salts were dissolved in aqueous HCl (0.16 M) where the mole fraction of Fe2+ to Fe3+ was adjusted to 0.5. This solution was dropped into 1 M aqueous NaOH solution (50 ml) at 80 °C under the inert atmosphere. After 30-min strong mechanical stirring, the magnetic particles were collected by centrifugation at 10,000 rpm for 15 min. The particles were washed three times with deionized water and dried at 50 °C under vacuum for 24 h.
⭒ Preparation of GO Nanosheets
Preparation of GO was accomplished by ultrasonication of graphite oxide powder. Graphite oxide solution was subjected to ultrasonication at room temperature for 1 h. The brown dispersion was then centrifuged at 3000 rpm for 30 min to remove any unexfoliated graphite oxide [40].
⭒ Preparation of GO/Fe3O4 Nanocomposite
The GO/Fe3O4 nanocomposite was synthesized by an ultrasonic-assisted reverse coprecipitation method, according to literature [41 ], with some modifications. The above-obtained GO (0.2 g) was added to distilled water (25 ml), and the mixture was subjected to ultrasonication at room temperature for 30 min. The iron salts, FeSO4.7H2O (0.48 g) and FeCl3.6H2O (0.47 g) were dissolved in 25 mL of distilled water, followed by the addition of NaOH (1 M) to the iron salts suspension to adjust pH 4. Then, GO aqueous dispersion was added into the above suspension. Again, NaOH (1 M) was added dropwise to adjust pH from 4 to 10 in order to obtain black precipitates. After that, the resulted suspension sonicated at 60 °C for 45 min. Finally, the obtained black GO/Fe3O4 nanoparticles were collected by magnetic separation, washed with water, and then dried under vacuum at 50 °C.
⭒ Activation of the Supports
In this procedure, 20 mg support material was dissolved in 5 ml of phosphate buffer (PBS) (50 mM, pH 7.0), including Triton X-100 or Tween 80 surfactants at five different concentrations (10, 20, 30, 40, 50 mM). The mixture was sonicated at room temperature for 45 min then the aggregates were separated and washed with deionized water three times. The solution of ethylenediamine (2 ml) in phosphate buffer (50 mM, pH 7.0) was added, and the suspension was stirred at room temperature for 2 h [42]. The precipitates were separated and washed with deionized water three times. Next, 250 mM of GLA or EPH (150, 200, 250, and 300 mM) as cross-linker was added to the mixture and shaken (150 rpm, 30 °C, 4 h). Finally, the activated support materials were recovered from the suspension, washed three times with deionized water and dried at room temperature for 24 h.
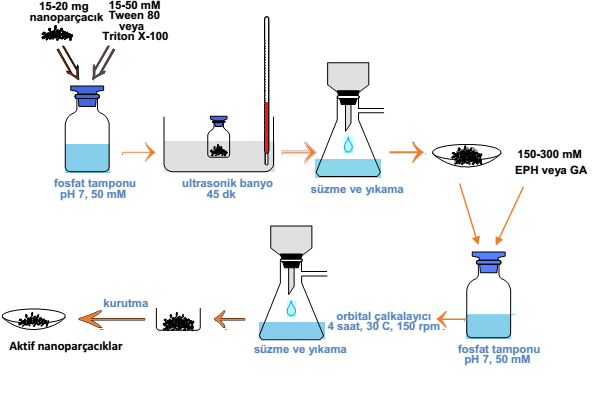
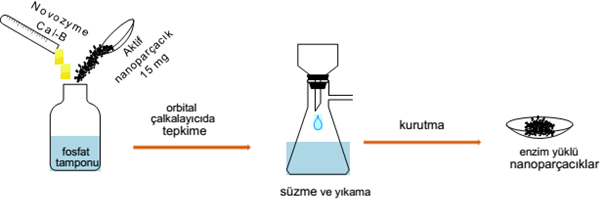
⭒ Lipase Immobilization onto Activated Nanoparticles
Activated nanoparticles soaked into phosphate buffer (5 ml, 50 mM) at different pH medium (pH 5, pH 6, pH 7, pH 8) including Cal-B (1 ml). The immobilization process was carried out in a thermal shaker (150 rpm), with different contact times (8, 12, 16, 20, 24 h) and immobilization temperatures (20, 30, 40, 50, 60 °C). Finally, the supports were washed three times with phosphate buffer (50 mM, pH 7) and dried at room temperature for 24 h.
⁎ Enzyme Activity Measurement
In order to investigate the enzyme activity, low acidity high refined olive oil was chosen as a substrate. In a conical flax, olive oil (2 ml), CaCl2 (0.1 M, 0.5 ml), phosphate buffer (0.07 M, pH 7, 3 ml), and deionized water (5 ml) were mixed and incubated in a thermal shaker (37 °C, 10 min, 150 rpm). After the incubation, native or immobilized lipase was added to the emulsion followed by reaction in a thermal shaker (37 °C, 20 min, 150 rpm). The acetoneethyl alcohol mixture (1:1 v/v) was added to the reaction medium to terminate the reaction. Titration method with NaOH (0.02 M) was chosen to measure the amount of released free fatty acid. A unit (U) lipase activity is known as the amount of a standard lipase preparation that releases 1 mol of fatty acid equivalent per minute from the substrate emulsion under certain assay conditions. To calculate the specific activity of both native and immobilized lipases, Eq. 1 was used [43].
Equation 1
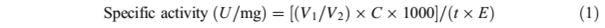
where V1 is the amount of consumed NaOH (for test) (ml), V2 is the amount of consumed NaOH (for blank) (ml), C is concentration of NaOH (mol/l), E is amount of the enzyme (mg), and t is incubation time (min).
⁎ Enantioselective Transesterif ication of 1-Phenyl Ethanol with Immobilized Lipases
For enzymatic kinetic resolution, vinyl acetate (240 mM for GO-GLA-CalB and GO/Fe3O4 - GLA-CalB, 480 mM for 3O4 -GLA-CalB) and rac-1-phenyl ethanol (120 mM) were dissolved in organic solvent n-hexane (total reaction volume is 3 ml) in screw-capped vials. GO-GLA-CalB (15.33 U/mg), 3O4-GLA-CalB (14.26 U/mg), or GO/3O4 -GLA-CalB (13.47 U/mg) and Applied Biochemistry and Biotechnology Author's personal copy molecular sieves (100 mg) were added to the mixture then stirred with constant shaking (150 rpm) at a fixed temperature (40 °C) in an orbital shaker. After the reaction, immobilized lipases were collected by filtering and organic solvent removed via rotary evaporator. The analysis of the samples was carried out by high-pressure liquid chromatography with a Chiral cell-OB column (4.6 mm × 50 mm, Daicel Chemical Ind. Ltd. France) using hexane: 2-propanol in the ratio of 95:05 at a flow rate of 0.90 ml/min and detected at 254 nm with a UV detector. The conversion (c) is described as fractional conversion [44]:
Equation 2
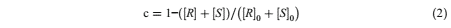
where [ R] is the concentration of (R)-1-phenyletehanol, [ S] is the concentration of (S)-1- phenyletehanol, [ R]0 is the initial concentration of (R)-1-phenyletehanol, and [ S]0 is the initial concentration of (S)-1-phenyletehanol. The enantiomeric excess (ee%) percent and the enantiomeric ratio (E) was calculated [1]:
Equation 3
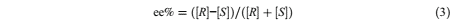
Equation 4
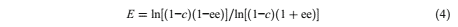
where c is conversion and ee% is the enantiomeric excess.